.webp)
What is a sequence alignment tool?
Imagine trying to find one misspelled word hidden across millions of books by scanning every page yourself. That is close to the problem scientists face when they work with DNA. The human genome has more than three billion letters, so reading it by hand is not realistic.
To handle that, researchers use sequence alignment software. It helps them compare long strings of A, T, C, and G and spot meaningful patterns fast. In simple terms, it works like a very fast search tool for genetic data.
Manual comparison is slow and full of room for error. With automated DNA alignment, computers can scan huge datasets in seconds. Work that once took years can now finish very quickly. That speed mattered during the COVID-19 pandemic, when scientists compared new viral sequences against known samples to trace mutations and track how variants were related. Modern biology depends on this kind of computation just as much as it depends on lab equipment.
Why do we need alignment tools? Solving the “corrupted text” puzzle
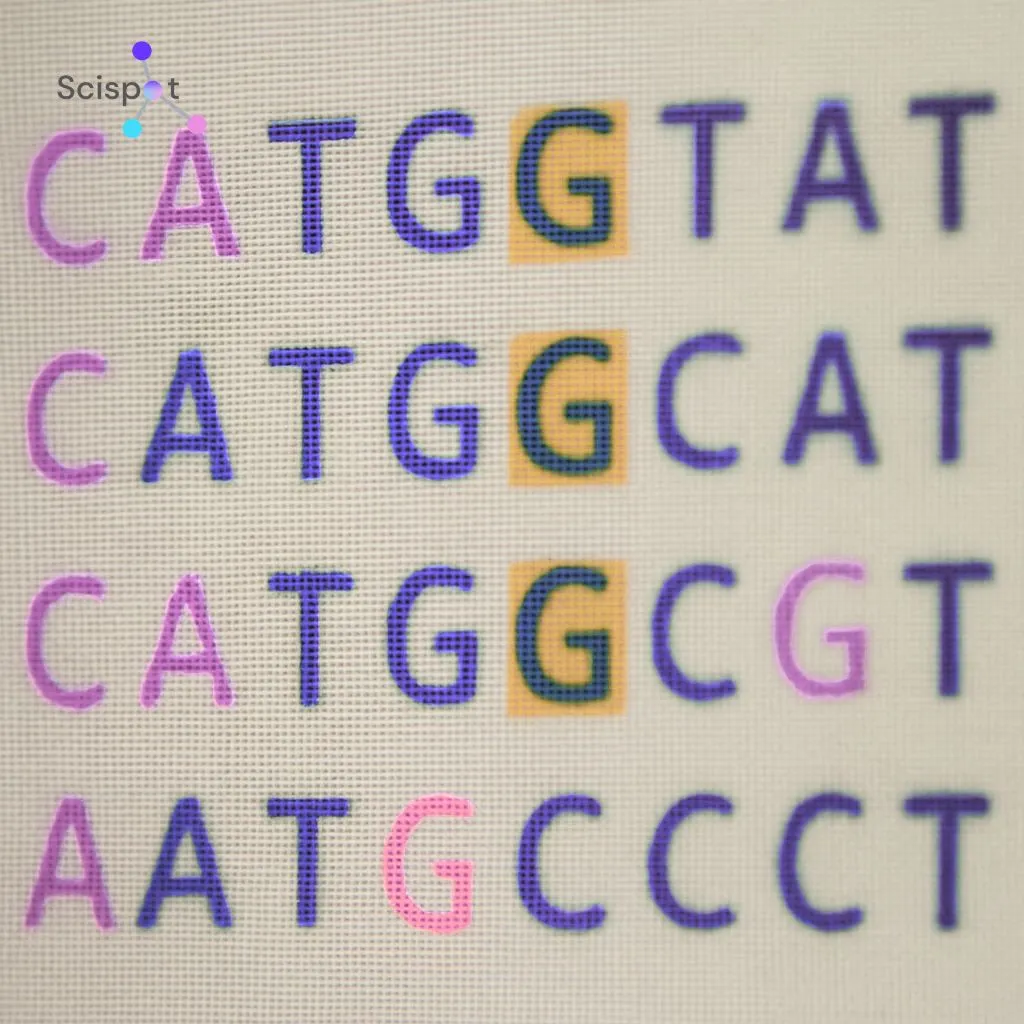
Think about sending a text message that arrives with a few letters missing or changed. You cannot compare the two versions line by line once one missing letter shifts everything after it. The same problem shows up in genetic code.
Over time, DNA picks up changes, or mutations. These usually show up in three forms:
- Mismatches: the wrong letter appears in the sequence
- Deletions: a letter is missing
- Insertions: an extra letter appears
Biologists often group insertions and deletions together as indels. Because of indels, two DNA strands can stop lining up after the first missing or extra letter. That makes direct comparison messy.
Sequence alignment software fixes this by adding blank spaces, called gaps, so matching letters line up again. To stop the program from forcing bad matches, it uses a gap penalty. That lowers the score each time a gap is added. By balancing matches against penalties, DNA sequence alignment helps researchers see which changes happened and where. That makes it useful for tracing virus evolution, studying ancient DNA, and comparing species.
Once that basic logic is in place, researchers still need to decide whether they want to compare whole sequences or just the part that matters.

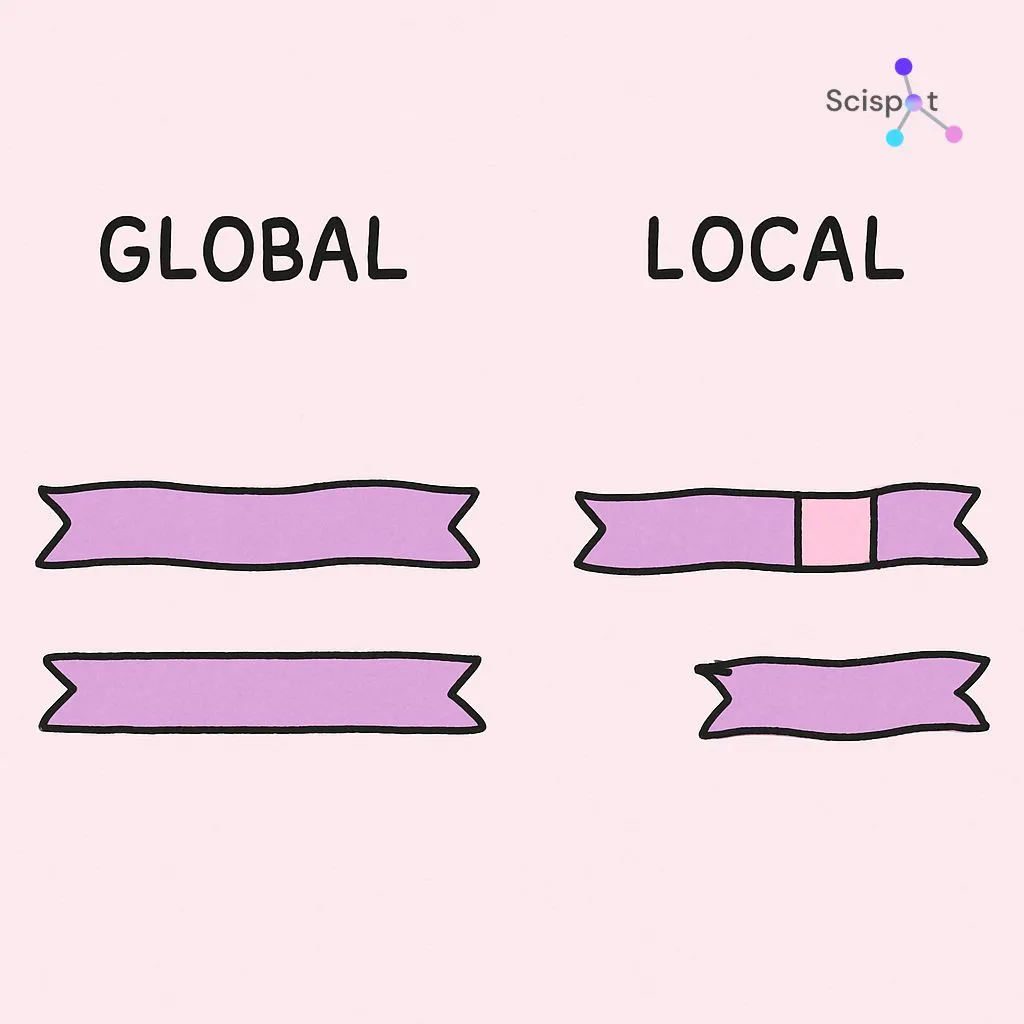
Global vs. local alignment: matching the whole blueprint or one room
Think of two building plans. Sometimes you want to know whether the full buildings match. Other times, you only care whether both contain the same kitchen design. Sequence alignment works the same way.
Global alignment compares two sequences from beginning to end. It forces every letter into the comparison, which makes sense when the sequences are already very similar, like two related genes. One well-known method for this is the Needleman-Wunsch algorithm, which scores similarity across the full length of both sequences.
But biology is often messier than that. A short DNA region can show up inside a much larger and mostly unrelated sequence. In that case, researchers use local alignment. This method looks for smaller regions of strong similarity and ignores the rest. The Smith-Waterman algorithm is a classic approach for this kind of search.
Because scientists often want to find one useful match inside a huge database, local alignment became a standard part of modern bioinformatics. Once that became common, the next problem was scale. There was simply too much data to compare one letter at a time.
How computers handle the heavy lifting: from BLAST to big data
Reading every sequence in a database one by one would take far too long. Databases like GenBank contain huge amounts of genetic data, so researchers use shortcuts called heuristics to speed up the search.
One of the best-known tools is BLAST. Instead of comparing everything against everything, BLAST looks for quick anchor points and builds from there. That cuts search time from something impractical to something usable.
BLAST usually works in three steps:
- Seeding: it looks for short exact matches
- Extension: it expands outward from those matches to see whether the pattern continues
- Evaluation: it scores the result to decide whether the match is likely to matter biologically
To judge whether a match is real or just random, scientists look at the E-value, or Expect value. This tells them how likely it is that the match happened by chance. Lower E-values suggest stronger confidence.
That final score depends on more than just matching letters. The software also needs rules for how different kinds of changes should count.

Scoring the game: how software measures genetic similarity
Sequence alignment depends on scoring. Exact matches earn points. Mismatches and gaps reduce the score. But not every mismatch means the same thing, especially in protein sequences.
To read these results well, scientists look at identity and similarity. Identity means the letters match exactly. Similarity means the letters may differ, but they still have a similar biological role.
To handle that, protein alignment software uses a substitution matrix. You can think of it as a scoring guide. Some changes are minor and cost very little. Others are more disruptive and carry a larger penalty. This helps researchers avoid treating every difference as equally important.
With that scoring system, sequence alignment becomes more than pattern matching. It becomes a way to measure biological relatedness in a structured way. Once those rules are clear, the next step is choosing the right tool for the job.
.webp)
Choosing your tool: Clustal Omega vs. MAFFT for large-scale data
Comparing two sequences is manageable. Comparing thousands at once is much harder. That task is called Multiple Sequence Alignment, or MSA.
The challenge gets bigger with proteins. DNA uses a four-letter alphabet. Proteins use twenty letters, so the number of possible comparisons rises fast. That is why researchers rely on specialized software.
Three common choices are:
- Clustal Omega: often chosen when accuracy matters most
- MAFFT: useful when speed is the priority for large datasets
- MEGA: a more visual option that is often easier for beginners to work with
Most labs use free, open-source tools like these rather than commercial platforms. Each tool has trade-offs. Clustal Omega is often trusted for accuracy. MAFFT is often preferred for speed. MEGA helps users explore results visually. The best choice depends on the size of the dataset and what the researcher needs to learn from it.

Bringing sequence alignment into one digital workflow with Scispot
Scispot fits naturally around sequence alignment work as the digital system that keeps everything connected. Tools like BLAST, Clustal Omega, and MAFFT do the alignment itself. Scispot helps labs manage the work around that process in one place.
Teams can centralize raw sequence files, link alignment outputs to samples and experiments, standardize review steps, track version history, and connect results to downstream analysis. That matters because sequence work often ends up spread across folders, spreadsheets, and separate software tools.
Instead of leaving that data scattered, Scispot gives labs a structured way to organize, trace, and use alignment results with less manual work. For teams that need more than a point tool, Scispot turns sequence alignment into part of a connected, audit-ready digital workflow.
From data to discovery: tracking outbreaks and building family trees
DNA is not just a string of letters. It is the input for software that helps power modern biology and medicine. By comparing sequences, scientists can build phylogenetic trees that show how organisms are related and how they changed over time.
As labs generate larger sequencing datasets, they also rely more on cloud-based systems to process and manage that data. These tools are already visible in public scientific resources. Researchers can run BLAST searches online, explore NCBI databases, and review variant tracking data to see how sequence comparison supports outbreak monitoring and evolutionary analysis.
Many major discoveries now depend on this work. They are not found through microscopes alone. They come from software that can align, score, and interpret genetic information in seconds.